Métabolisme
Le métabolisme est l'ensemble des réactions chimiques qui se déroulent au sein d'un être vivant pour lui permettre notamment de se maintenir en vie, de se reproduire, de se développer et de répondre aux stimuli de son environnement. Certaines de ces réactions chimiques se déroulent en dehors des cellules de l'organisme, comme la digestion ou le transport de substances entre cellules. Cependant, la plupart de ces réactions ont lieu dans les cellules elles-mêmes et constituent le métabolisme intermédiaire.
La biochimie cellulaire repose sur des réactions chimiques catalysées par des enzymes, c'est-à-dire des protéines possédant chacune la faculté de faciliter une réaction chimique spécifique. Ces réactions sont régies par les principes de la thermodynamique et s'organisent en voies métaboliques. Ces dernières sont constituées d'un ensemble de transformations permettant de convertir un composé chimique en un autre à travers des transformations successives, parallèles ou cycliques, catalysées par des enzymes. Certaines de ces enzymes sont soumises à une régulation par des métabolites cellulaires ou par des signaux extracellulaires. Ces facteurs de régulation modifient la cinétique enzymatique, accélérant ou ralentissant certaines réactions déterminantes, et aboutissant à l'autorégulation du système par l'ouverture et la fermeture des différentes voies métaboliques selon les circonstances.
Dans l'ensemble des réactions constituant le métabolisme, on distingue d'une part l'anabolisme, qui représente l'ensemble des voies de biosynthèse des constituants cellulaires, et d'autre part le catabolisme, qui représente l'ensemble des voies de dégradation de ces constituants cellulaires en petites molécules pour en libérer l'énergie par oxydation ou pour rebâtir d'autres constituants cellulaires. Les réactions de l'anabolisme et du catabolisme sont interconnectées à travers des molécules spécialisées jouant le rôle de cofacteurs enzymatiques. C'est par exemple le cas de l'adénosine triphosphate (ATP), dont l'hydrolyse en adénosine diphosphate (ADP) et en phosphate inorganique (Pi) est souvent couplée aux réactions d'anabolisme pour les rendre thermodynamiquement favorables. Le nicotinamide adénine dinucléotide (NAD+ à l'état oxydé) et le nicotinamide adénine dinucléotide phosphate (NADPH à l'état réduit), quant à eux, sont des transporteurs d'électrons utilisés dans les réactions d'oxydoréduction cellulaires, le NAD+ plutôt dans le catabolisme et le NADPH dans l'anabolisme. Des coenzymes permettent également d'échanger de la matière entre les différentes voies métaboliques. Ainsi, la coenzyme A permet d'activer des groupes acyle pour former une acyl-CoA, dont la plus importante est l'acétyl-CoA : cette dernière se trouve au carrefour de plusieurs voies métaboliques majeures, telles que la dégradation des glucides et des lipides, la production d'énergie métabolique, ou encore la biosynthèse des acides gras et des oses.
Le métabolisme d'un être vivant définit les types de substances chimiques qui sont des nutriments pour cet organisme et lesquels sont au contraire des poisons : ainsi, le sulfure d'hydrogène H2S est indispensable au développement de certains procaryotes, alors que ce gaz est toxique pour les animaux[1]. L'intensité du métabolisme de base détermine également la quantité de nourriture nécessaire à l'organisme.
Il est frappant d'observer la similitude des voies métaboliques fondamentales et des composés biochimiques à travers les organismes les plus divers[2]. Ainsi, les acides carboxyliques constituant les intermédiaires du cycle de Krebs se retrouvent chez tous les êtres vivants connus, allant d'un procaryote tel qu'E. coli jusqu'à un métazoaire tel que l'éléphant[3]. Ces similitudes remarquables sont très certainement dues à l'apparition précoce de ces voies métaboliques au cours de l'évolution des formes de vie sur Terre et à leur conservation en raison de leur efficacité[4],[5].
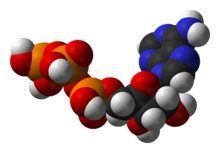
Molécule d'ATP, unité d'échange d'énergie universelle.

Molécule de NAD+, coenzyme des réactions d'oxydoréduction.

Molécule d'acétyl-CoA, coenzyme activant un résidu acétyle.
Sommaire
1 Composés biochimiques fondamentaux
1.1 Acides aminés et protéines
1.2 Lipides
1.3 Glucides
1.4 Nucléotides
1.5 Coenzymes et cofacteurs
1.6 Sels minéraux
2 Catabolisme
2.1 Digestion
2.2 Libération de l'énergie des composés organiques
3 Énergie et métabolisme
3.1 Phosphorylation oxydative
3.2 Libération de l'énergie des composés inorganiques
3.3 Absorption de l'énergie lumineuse
4 Anabolisme
4.1 Fixation du carbone
4.2 Glucides et glycanes
4.3 Acides gras, terpénoïdes et stéroïdes
4.4 Protéines
4.5 Voie de novo et voie de sauvetage des nucléotides
5 Xénobiotiques et stress oxydant
6 Régulation et contrôle du métabolisme
7 Évolution
8 Thermodynamique du métabolisme
9 Exemples de réactions métaboliques
10 Métabolisme et température
11 Métabonomique
12 Autre contenu sémantique
13 Notes et références
14 Annexes
14.1 Articles connexes
14.2 Bibliographie
14.3 Liens externes
Composés biochimiques fondamentaux |

Molécule de triglycéride, constituant principal de la graisse.
Les animaux, les plantes et les microbes sont formés de trois grandes familles de molécules :
les lipides, qui jouent un rôle à la fois de réserve d'énergie, de constituant principal des membranes de leurs cellules, et de communication entre cellules par des mécanismes de signalisation lipidique ;
les peptides, qui jouent un rôle déterminant à la fois dans la structure des organismes (protéines), leur biochimie (enzymes) et l'intégration physiologique entre les organes (hormones peptidiques) ;
les glucides, qui servent à la fois à stocker de l'énergie, à stabiliser certaines protéines et à favoriser l'adhérence des cellules entre elles, par exemple dans les mécanismes de reconnaissance du système immunitaire à travers les lectines.
Ces molécules étant essentielles à la vie, le métabolisme cellulaire consiste ou bien à les synthétiser pour produire de nouvelles cellules et faire croître les tissus, ou bien à les dégrader lors de la digestion pour les utiliser comme sources d'énergie et de constituants élémentaires qui peuvent être recyclés dans la biosynthèse de nouvelles biomolécules.
Les macromolécules biologiques sont elles-mêmes des polymères appartenant à trois familles différentes :
les polypeptides, qui sont constitués d'acides aminés, au sein desquels on trouve les protéines et les enzymes ;
les polysaccharides, qui sont constitués d'oses — par exemple l'amidon, la cellulose, le glycogène ;
les polynucléotides, qui sont constitués de nucléotides, et dont les deux représentants sont les acides ribonucléiques (ARN) et les acides désoxyribonucléiques (ADN), lesquels portent le code génétique, qui détermine notamment la nature des protéines et des enzymes — et donc la physiologie — de chaque cellule.
Acides aminés et protéines |
Les protéines sont constituées d'acides α-aminés liés entre eux par une liaison peptidique pour former une chaîne linéaire. De nombreuses protéines sont des enzymes qui catalysent des réactions chimiques du métabolisme. D'autres protéines ont un rôle structurel ou mécanique, comme celles du cytosquelette, qui maintient la forme générale de la cellule[6]. Les protéines jouent également un rôle clé dans la signalisation cellulaire, comme anticorps du système immunitaire, l'adhérence cellulaire, le transport actif à travers les membranes et le cycle cellulaire. Les acides aminés contribuent également à fournir de l'énergie au métabolisme cellulaire en alimentant le cycle de Krebs[7], en particulier lorsque les principales sources d'énergie, telles que le glucose, font défaut, ou lorsque la cellule est soumise à un stress métabolique[8].
Lipides |
Les lipides sont le groupe de composés biochimiques le plus diversifié. Leur fonction structurelle principale est celle de constituant des membranes cellulaires, notamment de la membrane plasmique et du système endomembranaire des cellules eucaryotes, ainsi que de celles d'organites telles que les mitochondries et les chloroplastes, voire de sous-organites tels que les thylakoïdes. Ils sont également utilisés comme sources d'énergie. On les définit généralement comme des molécules biologiques hydrophobes et amphiphiles, solubles dans les solvants organiques tels que le benzène et le chloroforme[9]. Les graisses sont, parmi les lipides, un grand groupe de composés solides essentiellement constitués d'acides gras et de glycérol. Une molécule formée de trois résidus d'acides gras estérifiant les trois hydroxyles d'un résidu de glycérol est appelée triglycéride. Il existe diverses variations autour de ce thème central, par exemple avec de la sphingosine dans le cas des sphingolipides, et des groupes hydrophiles tels que le groupe phosphate dans le cas des phospholipides. Les stéroïdes, tels que le cholestérol, sont une autre famille importante de lipides[10].
Glucides |

Représentations du glucose, de la projection de Fischer (linéaire) à la projection de Haworth (cyclique).
Les glucides sont des aldéhydes ou des cétones ayant plusieurs groupes hydroxyle. Ces molécules peuvent exister sous forme linéaire ou cyclique. Ce sont les molécules biologiques les plus abondantes. Elles remplissent un grand nombre de fonctions, comme substances de stockage et le transport de l'énergie (amidon, glycogène) ou comme composants structurels (cellulose chez les plantes, chitine chez les animaux). Les monomères glucidiques sont appelés oses : ce sont par exemple le galactose, le fructose, et surtout le glucose. Ils peuvent polymériser en donnant des polysaccharides avec une variété de structures quasiment infinie[11].
Nucléotides |
Les nucléosides résultent de la liaison d'une molécule de ribose ou de désoxyribose à une base nucléique. Ces dernières sont des composés hétérocycliques contenant des atomes d'azote ; elles se divisent en purines et pyrimidines. Les nucléotides sont formés d'un nucléoside et d'un ou plusieurs groupements phosphates liés au sucre.
Les deux acides nucléiques, l'acide ribonucléique (ARN) et l'acide désoxyribonucléique (ADN), sont des polymères de nucléotides, ou polynucléotides. L'ARN est constitué de ribonucléotides (contenant un ribose) et l'ADN de désoxyribonucléotides (contenant un désoxyribose). Les acides nucléiques permettent le codage et l'expression de l'information génétique ainsi que son décodage à travers les processus successifs de transcription et de traduction génétique de la biosynthèse des protéines. Cette information est préservée par les mécanismes de réparation de l'ADN et transmise à travers le processus de réplication de l'ADN. De nombreux virus, dits virus à ARN, ont un génome constitué d'ARN et non d'ADN — c'est par exemple le cas du virus de l'immunodéficience humaine (VIH) ou du virus de la grippe— certains ont recours à des transcriptases inverses pour générer dans la cellule hôte une matrice d'ADN à partir du génome viral en ARN[12], d'autres sont directement répliqués d'ARN en ARN par une ARN polymérase-ARN dépendante (ou réplicase). L'ARN des ribozymes, tels que les spliceosomes (ou particules d'épissage) et les ribosomes, est semblable aux enzymes dans la mesure où il est capable de catalyser des réactions chimiques.
Coenzymes et cofacteurs |
Le métabolisme implique un très grand nombre de réactions chimiques différentes formant un réseau de transformations complexe, mais la plupart d'entre elles peuvent être rapprochées de quelques types de réactions basiques consistant en des transferts de groupes fonctionnels[13]. Cela résulte du fait que la biochimie cellulaire fait appel à un nombre relativement restreint de molécules agissant comme des activateurs susceptibles de transporter des groupes d'atomes entre différentes réactions[14]. De telles molécules sont appelées coenzymes. Chaque type de transfert de groupe fonctionnel fait appel à une coenzyme spécifique. Chacune de ces coenzymes est également spécifique d'un certain nombre d'enzymes qui catalysent les réactions de transfert, enzymes qui les altèrent et les régénèrent en permanence[15].

(en) La succinate déshydrogénase fait intervenir plusieurs cofacteurs, dont un FADH2, des centres fer-soufre et un hème.
L'adénosine triphosphate (ATP) est la coenzyme universelle des échanges d'énergie chez tous les organismes connus. Ce nucléotide permet de transférer de l'énergie métabolique entre les réactions qui libèrent de l'énergie et celles qui en absorbent. Il n'y a à chaque instant qu'une faible quantité d'ATP dans les cellules, mais, comme ce capital d'ATP est continuellement consommé et régénéré, le corps humain peut en réalité consommer chaque jour une masse d'ATP pratiquement équivalente à son poids total[15]. L'ATP permet de coupler l'anabolisme au catabolisme, le premier consommant l'ATP produit par le second. Il sert également de transporteur de groupes phosphate dans les réactions de phosphorylation.
Les vitamines sont des composés organiques indispensables en petite quantité au fonctionnement des cellules mais que ces dernières ne peuvent pas produire elles-mêmes. Chez l'homme, la plupart des vitamines deviennent des coenzymes après quelques transformations dans les cellules. Ainsi, les vitamines hydrosolubles (vitamines B) sont phosphorylées ou couplées à des nucléotides lorsqu'elles sont utilisées dans les cellules. Par exemple, la niacine (acide nicotinique) entre dans la composition du nicotinamide adénine dinucléotide (NAD+) et du nicotinamide adénine dinucléotide phosphate (NADP+), qui sont des coenzymes importantes impliquées dans les réactions d'oxydoréduction comme accepteurs d'hydrogène. Il existe des centaines de déshydrogénases, qui soustraient des électrons de leur substrat et réduisent le NAD+ en NADH et H+. Cette forme réduite de la coenzyme peut alors être utilisée par une réductase[16]. Le couple NAD+ / NADH intervient davantage dans les réactions cataboliques tandis que le couple NADP+ / NADPH est spécifique à l'anabolisme.
Sels minéraux |
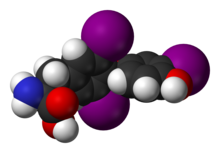
Molécule de triiodothyronine, principale hormone thyroïdienne, contenant trois atomes d'iode (ici en violet).
Les sels minéraux jouent un rôle déterminant dans le métabolisme. Certains sont abondants, comme le sodium et le potassium, tandis que d'autres ne sont actifs qu'à faible concentration. Environ 99 % de la masse des mammifères est constituée des éléments carbone, azote, calcium, sodium, chlore, potassium, hydrogène, phosphore, oxygène et soufre[17]. Les composés organiques (protéines, lipides et glucides) contiennent l'essentiel du carbone et de l'azote, tandis que l'essentiel de l'oxygène et de l'hydrogène sont présents sous forme d'eau.
Les sels minéraux les plus abondants agissent comme électrolytes. Les principaux ions sont le sodium Na+, le potassium K+, le calcium Ca2+, le magnésium Mg2+, le chlorure Cl–, le phosphate PO43− et l'ion organique bicarbonate HCO3–. Le maintien de gradients de concentration déterminés à travers les membranes cellulaires permet de maintenir l'équilibre osmotique et le pH du milieu intracellulaire. Les ions sont également essentiels au fonctionnement des nerfs et des muscles grâce au potentiel d'action issu de l'échange d'ions, à travers la membrane plasmique, entre le fluide extracellulaire (en) et le fluide intracellulaire, c'est-à-dire le cytosol. Les ions entrent et quittent les cellules en empruntant des protéines membranaires appelées canaux ioniques. Ainsi, la contraction musculaire dépend du passage des ions calcium, sodium et potassium à travers les canaux ioniques de la membrane cellulaire et les tubules T[18].
Les métaux de transition sont généralement présents à l'état de trace chez les organismes vivants, le zinc et le fer étant les plus abondants d'entre eux[19],[20]. Ces métaux interviennent comme cofacteurs de certaines protéines et enzymes et sont essentiels à leur bon fonctionnement. C'est par exemple le cas d'une enzyme telle que la catalase et d'une protéine transporteuse d'oxygène telle que l'hémoglobine[21]. Les cofacteurs métalliques se lient spécifiquement à certains sites des protéines. Bien que les cofacteurs puissent être modifiés au cours de la réaction catalysée, ils reviennent toujours à leur état d'origine à la fin de la réaction. Ils sont absorbés par les organismes à l'aide de transporteurs spécifiques, par exemple les sidérophores pour absorber le fer, et sont liés à des protéines de stockage telles que la ferritine et les métallothionéines lorsqu'ils ne sont pas utilisés[22],[23].
Catabolisme |
Le catabolisme est l'ensemble des processus métaboliques de dégradation des biomolécules. Cela comprend par exemple la dégradation et l'oxydation des nutriments. Le catabolisme a pour fonction de fournir l'énergie et les constituants élémentaires indispensables au métabolisme de la cellule. La nature exacte de ces réactions dépend de chaque organisme. Les êtres vivants peuvent être classés en fonction de leur sources d'énergie et de carbone, ce qu'on appelle leur type trophique :
| Source d'énergie | Lumière solaire | photo- | -trophe | ||
| Composés chimiques | chimio- | ||||
| Donneur d'électrons | Composés organiques | organo- | | ||
| Composés inorganiques | litho- | ||||
| Source de carbone | Composés organiques | hétéro- | |||
| Composés inorganiques | auto- | ||||
Les organotrophes utilisent des molécules organiques comme source d'énergie tandis que les lithotrophes utilisent des substrats inorganiques et que les phototrophes convertissent l'énergie solaire en énergie chimique. Ces différents métabolismes reposent cependant tous sur le transfert d'électrons de composés donneurs — tels que des molécules organiques, l'eau, l'ammoniac, le sulfure d'hydrogène ou encore des cations de fer(II) Fe2+ (fer ferreux) — vers des composés accepteurs d'électrons tels que l'oxygène, les nitrates ou encore les sulfates[24]. Chez les animaux, ces réactions conduisent à dégrader des molécules organiques complexes en molécules plus simples telles que le dioxyde de carbone et l'eau. Chez les organismes photosynthétiques tels que les plantes et les cyanobactéries, ces réactions permettent de libérer l'énergie de la lumière solaire absorbée et stockée par l'organisme.
Les principaux groupes de réactions cataboliques chez les animaux peuvent être classés en trois étapes principales. Dans la première, les grandes molécules organiques telles que les protéines, les polysaccharides ou les lipides sont digérés en leurs composants élémentaires à l'extérieur des cellules. Puis ces composants élémentaires sont absorbés par les cellules et convertis en métabolites encore plus petits, le plus souvent en acétyl-coenzyme A (acétyl-CoA), avec libération d'un peu d'énergie. Enfin, le résidu acétyle de l'acétyl-CoA est oxydé en eau et dioxyde de carbone par le cycle de Krebs et la chaîne respiratoire, cette dernière permettant de libérer l'énergie des électrons à haut potentiel transférés au NADH au cours du cycle de Krebs.
Digestion |
Les macromolécules telles que l'amidon, la cellulose et les protéines, qui sont des biopolymères, ne peuvent être absorbées facilement par les cellules et doivent être clivées en oligomères, voire en monomères, afin de pouvoir être métabolisées. C'est ce qu'on appelle la digestion. Plusieurs classes d'enzymes communes réalisent ces transformations, par exemple les peptidases, qui clivent les protéines en oligopeptides et en acides aminés, ou encore les glycoside hydrolases (ou glycosidases), qui clivent les polysaccharides en oligosaccharides et en oses.
Les microorganismes sécrètent leurs enzymes digestives dans leur voisinage[25],[26] alors que les animaux sécrètent ces enzymes uniquement à partir de cellules spécialisées situées dans leur appareil digestif. Les acides aminés et les oses libérés par ces enzymes extracellulaires sont ensuite absorbées à travers la membrane plasmique des cellules par des protéines membranaires de transport actif[27],[28].
Libération de l'énergie des composés organiques |

Cycle de Krebs et cycle du glyoxylate avec leurs métabolites (acétyl-CoA, citrate, cis-aconitate, isocitrate, α-cétoglutarate, succinyl-CoA, succinate, fumarate, malate, oxaloacétate et glyoxylate) et leurs enzymes (citrate synthase, aconitase, isocitrate déshydrogénase, complexe alpha-cétoglutarate déshydrogénase, succinyl-CoA synthétase, succinate déshydrogénase, fumarase, malate déshydrogénase, isocitrate lyase et malate synthase).
Les glucides sont généralement absorbés par les cellules après avoir été préalablement digérés en oses[29]. La principale voie de dégradation des oses à l'intérieur de la cellule est la glycolyse, qui produit quelques molécules d'ATP et deux molécules de pyruvate par molécule de glucose dégradée[30]. Le pyruvate est un métabolite commun à plusieurs voies métaboliques, mais l'essentiel est converti en acétyl-CoA pour alimenter le cycle de Krebs. Ce dernier produit encore quelques molécules d'ATP, mais son produit essentiel est le NADH, issu de la réduction du NAD+ lors de l'oxydation de l'acétyl-CoA. Cette oxydation libère du dioxyde de carbone comme sous-produit. En conditions anaérobies, la glycolyse produit du lactate par transfert des électrons du NADH au pyruvate par une lactate déshydrogénase en vue de régénérer du NAD+ pour la glycolyse. Une voie alternative pour la dégradation du glucose est la voie des pentoses phosphates, qui a pour fonction première non pas de libérer de l'énergie, mais de produire des précurseurs de diverses biosynthèses tels que du NADPH, utilisé notamment pour la biosynthèse des acides gras, ainsi que du ribose-5-phosphate, utilisé pour la synthèse des nucléotides, et de l'érythrose-4-phosphate, précurseur d'acides aminés aromatiques.
Les lipides sont dégradés par hydrolyse en glycérol et acides gras. Le glycérol est dégradé par la glycolyse tandis que les acides gras le sont par la β-oxydation pour produire de l'acétyl-CoA, dégradé à son tour par le cycle de Krebs. L'oxydation des acides gras libère davantage d'énergie que les glucides car ces derniers contiennent plus d'oxygène et sont donc davantage oxydés que les acides gras.
Les acides aminés sont utilisés ou bien pour produire des protéines et diverses autres biomolécules, ou bien oxydés en urée et dioxyde de carbone pour libérer de l'énergie[31]. Leur oxydation commence par leur conversion en α-cétoacide par une transaminase qui clive leur groupe amine, ce dernier alimentant le cycle de l'urée. Plusieurs de ces α-cétoacides sont des intermédiaires du cycle de Krebs : la désamination du glutamate donne ainsi de l'α-cétoglutarate[32]. Les acides aminés glucoformateurs peuvent également être convertis en glucose à travers la néoglucogenèse[33].
Énergie et métabolisme |
Phosphorylation oxydative |
Au cours de la phosphorylation oxydative — qu'il faudrait appeler plus correctement en français oxydation phosphorylante — les électrons à haut potentiel, issus des réactions d'oxydation du métabolisme, sont transférés à de l'oxygène avec libération d'énergie, cette énergie étant récupérée pour synthétiser de l'ATP. Ceci est réalisé par les eucaryotes à travers une série de protéines membranaires des mitochondries formant la chaîne respiratoire. Chez les procaryotes, ces protéines se trouvent dans la membrane interne[34]. Ces protéines membranaires utilisent l'énergie libérée par la circulation des électrons depuis les coenzymes réduites telles que le NADH et le FADH2 vers l'oxygène pour pomper des protons à travers la membrane mitochondriale interne (chez les eucaryotes) ou la membrane plasmique (chez les procaryotes)[35].
Fonctionnement de l'ATP synthase illustrant la phosphorylation de l'ADP en ATP.
Le pompage des protons hors de la matrice mitochondriale ou du cytoplasme génère un gradient de concentration de protons à travers les membranes — c'est-à-dire une différence de pH. Il en découle un gradient électrochimique[36]. Cette « force proton motrice » actionne une enzyme appelée ATP synthase qui fonctionne comme une turbine qui catalyse la phosphorylation de l'ADP en ATP au passage des protons qui retournent vers la matrice mitochondriale à travers la membrane mitochondriale interne[15].
Libération de l'énergie des composés inorganiques |
La chimiolithotrophie (en) est un type trophique définissant les procaryotes qui tirent leur énergie de composés inorganiques. Ces organismes peuvent utiliser l'hydrogène[37], les composés réduits du soufre[1] — sulfure S2–, sulfure d'hydrogène H2S, thiosulfate S2O32− — le fer ferreux (Fe2+)[38] et l'ammoniac (NH3)[39] comme donneurs d'électrons qu'ils transfèrent à des accepteurs tels que l'oxygène O2 ou l'anion nitrite (NO2–)[40]. Ces processus microbiens sont importants du point des vue des cycles biogéochimiques planétaires tels que le cycle de l'azote, la nitrification et la dénitrification, et sont déterminants pour la fertilité des sols[41],[42].
Absorption de l'énergie lumineuse |

Couplage chimiosmotique entre l'énergie lumineuse, la bactériorhodopsine et l'ATP synthase chez Halobacterium salinarum, une archée halophile aérobie. La paroi cellulaire n'est pas représentée.
L'énergie lumineuse est absorbée par les plantes, les cyanobactéries, les bactéries pourpres, les bactéries vertes sulfureuses et certains protistes. Ce processus est souvent couplé à la conversion du dioxyde de carbone en composés organiques dans le cadre de la photosynthèse. Ces deux processus — absorption de l'énergie lumineuse et biosynthèse de composés organiques — peuvent néanmoins fonctionner séparément chez les procaryotes. Ainsi, les bactéries pourpres et les bactéries vertes sulfureuses peuvent utiliser la lumière du soleil comme source d'énergie et en même temps mettre en œuvre ou bien un processus de fixation du carbone ou bien un processus de fermentation des composés organiques[43],[44].
Chez de nombreux organismes, l'absorption de l'énergie solaire repose sur des principes semblables à ceux de la phosphorylation oxydative dans la mesure où un phénomène physique — la récupération de l'énergie des électrons de coenzymes réduites — est couplé à un phénomène chimique — la phosphorylation de l'ADP en ATP — par chimiosmose au moyen d'un gradient de concentration de protons générant un gradient électrochimique à travers une membrane[15]. Dans le cas de la photosynthèse, les électrons à haut potentiel sont issus de protéines d'absorption de l'énergie lumineuse appelées centres réactionnels photosynthétiques ou rhodopsines. Les centres réactionnels se déclinent en deux photosystèmes selon le pigment photosynthétique présent : la plupart des bactéries photosynthétiques n'en ont qu'un, tandis que les plantes et les cyanobactéries en ont deux[45].
Chez les plantes, les algues et les cyanobactéries, le photosystème II transfère l'énergie lumineuse à deux électrons d'une molécule d'eau qui sont captés par le complexe cytochrome b6f tandis que de l'oxygène O2 est libéré. L'énergie des électrons à haut potentiel transférés au complexe cytochrome b6f est utilisée pour pomper des protons à travers les membranes des thylakoïdes dans les chloroplastes, protons dont le retour dans le lumen s'accompagne de la phosphorylation d'ADP en ATP par une ATP synthase, comme dans le cas de la phosphorylation oxydative. Les électrons passent ensuite à travers le photosystème I et peuvent réduire une coenzyme NADP+ en NADPH en vue de son utilisation par le cycle de Calvin, ou bien être utilisés pour produire encore davantage d'ATP[46].
Anabolisme |
L'anabolisme comprend l'ensemble des voies métaboliques qui utilisent l'énergie (ATP) et le pouvoir réducteur (NADH) produits par le catabolisme pour synthétiser des biomolécules complexes. De manière générale, les molécules complexes qui contribuent aux structures cellulaires sont construites étape par étape à partir de précurseurs bien plus petits et plus simples.
L'anabolisme comprend trois étapes principales :
- tout d'abord la production de précurseurs tels que les acides aminés, les oses, les isoprénoïdes et les nucléotides ;
- puis leur activation sous une forme réactive du point de vue biochimique en utilisant l'énergie de l'ATP ;
- enfin l'assemblage de ces précurseurs activés pour construire des molécules complexes telles que les protéines, les polysaccharides, les lipides et les acides nucléiques.
Les organismes diffèrent dans le nombre des constituants de leurs cellules qu'ils sont capables de produire eux-mêmes. Les autotrophes tels que les plantes peuvent synthétiser les molécules organiques complexes de leurs cellules tels que les polysaccharides et les protéines à partir de molécules très simples comme le dioxyde de carbone CO2 et l'eau H2O. En revanche, pour produire leurs biomolécules complexes, les hétérotrophes ont besoin de nutriments plus complexes comme des sucres et des acides aminés. Les organismes peuvent être classés plus finement en fonction de leur source d'énergie première : les photoautotrophes et les photohétérotrophes tirent leur énergie de la lumière du soleil tandis que les chimioautotrophes et les chimiohétérotrophes tirent leur énergie de réactions d'oxydoréduction.
Fixation du carbone |

cellules d'une plante délimitées par une paroi de forme vaguement hexagonale et remplies de chloroplastes (en vert), qui sont le siège de la photosynthèse.
La photosynthèse est la biosynthèse de glucides à partir d'eau et de dioxyde de carbone en utilisant la lumière du soleil. Chez les plantes, les algues et les cyanobactéries, la molécule d'eau H2O est scindée en oxygène O2 et en électrons à haut potentiel dont l'énergie est utilisée pour phosphoryler de l'ADP en ATP et pour former du NADPH utilisé pour réduire le dioxyde de carbone en 3-phosphoglycérate, lui-même précurseur du glucose. Cette réaction de fixation du carbone est réalisée par la Rubisco, une enzyme essentielle du cycle de Calvin[47]. Il existe trois types différents de photosynthèse chez les plantes : la fixation du carbone en C3, la fixation du carbone en C4 et le métabolisme acide crassulacéen (CAM). Ces types de réactions diffèrent par la voie empruntée par le dioxyde de carbone pour entrer dans le cycle de Calvin : les plantes en C3 le fixent directement, tandis que les plantes en C4 et à photosynthèse CAM fixent le CO2 préalablement sur un autre composé comme adaptation aux températures élevées et aux conditions arides[48].
Chez les procaryotes photosynthétiques, les mécanismes de fixation du carbone sont plus diversifiés. Ce processus peut être réalisé par le cycle de Calvin, mais aussi par un cycle de Krebs inverse[49] ou par carboxylation de l'acétyl-CoA[50],[51]. Les organismes chimioautotrophes procaryotiques fixent également le carbone du CO2 en utilisant le cycle de Calvin mais avec de l'énergie provenant de l'oxydation de composés inorganiques[52].
Glucides et glycanes |
Au cours de l'anabolisme des glucides, des acides organiques simples peuvent être convertis en oses tels que le glucose, puis être polymérisés en polysaccharides tels que l'amidon. La biosynthèse du glucose à partir de composés tels que pyruvate, lactate, glycérol, 3-phosphoglycérate et acides aminés est appelée néoglucogenèse. La néoglucogenèse convertit le pyruvate en glucose-6-phosphate en passant par une série de métabolites dont de nombreux sont également des intermédiaires de la glycolyse[30]. Cependant, cette voie métabolique ne doit pas être vue comme la glycolyse prise en sens inverse car plusieurs de ses étapes sont catalysées par des enzymes différentes de la glycolyse. Ce point est important car il permet de réguler la biosynthèse et la dégradation du glucose de façon distincte l'une de l'autre et donc d'empêcher de voir ces deux processus fonctionner en même temps, l'un détruisant l'autre en pure perte[53],[54].
Bien que les organismes stockent couramment l'énergie sous forme de lipides, les vertébrés tels que les humains ne peuvent convertir les acides gras de leurs graisses en glucose au moyen de la néoglucogenèse car ils ne peuvent pas convertir l'acétyl-CoA en pyruvate : les plantes disposent de l'équipement enzymatique nécessaire pour ce faire, mais pas les animaux[55]. En conséquence, les vertébrés soumis à un jeûne prolongé utilisent leurs lipides pour produire des corps cétoniques destinés à pallier le manque de glucose dans les cellules qui ne sont pas en mesure de dégrader les acides gras pour produire leur énergie, notamment les cellules du cerveau[56]. D'autres organismes, tels que les plantes et les bactéries, traitent ce stress métabolique à l'aide du cycle du glyoxylate, qui court-circuite l'étape de décarboxylation du cycle de Krebs et permet la transformation de l'acétyl-CoA en oxaloacétate, ce dernier pouvant alors être utilisé pour produire du glucose[55],[57].
Les polysaccharides et les glycanes sont produits par addition séquentielle d'oses par une glycosyltransférase à partir d'un donneur ose-phosphate tel que l'uridine diphosphate glucose (UDP-glucose) sur un groupe hydroxyle accepteur d'un polysaccharide en cours de biosynthèse. Comme chacun des groupes hydroxyle du substrat peut être accepteur, les polysaccharides peuvent être linéaires ou ramifiés[58]. Les polysaccharides produits peuvent avoir un rôle structurel ou métabolique en eux-mêmes, ou bien encore être transférés à des lipides ou à des protéines par des enzymes appelées oligosaccharyltransférases[59],[60].
Acides gras, terpénoïdes et stéroïdes |
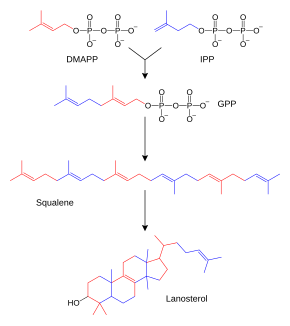
Représentation simplifiée de la biosynthèse des stéroïdes. Pour une meilleure lisibilité, seuls les intermédiaires principaux sont représentés : diméthylallyl-pyrophosphate (DMAPP), isopentényl-pyrophosphate (IPP), géranyl-pyrophosphate (GPP), squalène et lanostérol.
Les acides gras sont synthétisés par l'acide gras synthase (FAS), un ensemble d'enzymes qui catalyse la condensation de Claisen d'unités malonyl-CoA sur une amorce d'acétyl-CoA. Les chaînes acyle sont allongées par une séquence de quatre réactions qui se reproduisent en boucle à l'occasion de chaque condensation d'une nouvelle unité malonyl-CoA. Chez les animaux et les mycètes (champignons), ces réactions sont réalisées par un complexe enzymatique multifonctionnel appelé FAS I[61], tandis que chez les plantes et les bactéries ces réactions sont catalysées par un ensemble d'enzymes distinctes appelé FAS II dont chaque enzyme est monofonctionnelle[62],[63].
Les terpènes et terpénoïdes forment une grande famille de lipides qui comprennent les caroténoïdes et constituent la principale classe de produits naturels des plantes[64]. Ces composés résultent de l'assemblage et de la modification d'unités isoprène issues de précurseurs réactifs tels que l'isopentényl-pyrophosphate et le diméthylallyl-pyrophosphate[65]. Ces précurseurs peuvent être produits de différentes façons. Chez les animaux et les archées, la voie du mévalonate les synthétise à partir de l'acétyl-CoA[66] tandis que chez les plantes et les bactéries la voie du méthylérythritol phosphate, dite également voie non mévalonique par anglicisme, les produit à partir du pyruvate et du 3-phosphoglycérate[65],[67]. Ces donneurs d'unités isoprène sont utilisés notamment dans la biosynthèse des stéroïdes, tout d'abord pour former du squalène, qui est ensuite replié pour faire apparaître des cycles constituants du lanostérol[68]. Ce stérol peut alors être converti en d'autres stéroïdes tels que le cholestérol et l'ergostérol[68],[69].
Protéines |
Les organismes possèdent des capacités très variables de synthétiser les 22 acides aminés protéinogènes. La plupart des bactéries et des plantes peuvent produire tous ceux dont ils ont besoin, mais les mammifères ne peuvent synthétiser eux-mêmes que douze acides aminés, dits non essentiels, ce qui signifie que leur alimentation doit leur en apporter neuf autres : histidine, isoleucine, leucine, lysine, méthionine, phénylalanine, thréonine, tryptophane et valine — ils n'utilisent pas la pyrrolysine, spécifique aux archées méthanogènes.
Certains organismes simples, tels que la bactérie Mycoplasma pneumoniae, sont incapables de synthétiser le moindre acide aminé et les absorbent tous à partir de leur hôte[70]. Tous les acides aminés sont synthétisés à partir d'intermédiaires de la glycolyse, du cycle de Krebs et de la voie des pentoses phosphates. L'azote provient du glutamate et de la glutamine. La synthèse des acides aminés dépend de la formation de l'α-cétoacide approprié, qui est alors transaminé pour former l'acide aminé.
Les acides aminés sont assemblés en protéines en formant entre eux des liaisons peptidiques aboutissant à des chaînes linéaires polypeptidiques. Chaque protéine a une séquence déterminée en résidus d'acides aminés : c'est leur structure primaire. Les acides aminés peuvent s'associer en un nombre pratiquement illimité de combinaisons différentes, chaque combinaison correspondant à une protéine particulière. Les protéines sont assemblées à partir d'acides aminés qui sont préalablement activés sur une molécule d'ARN de transfert (ARNt) par une liaison ester. Ce précurseur, appelé aminoacyl-ARNt, est formé sous l'action d'enzymes spécifiques, les aminoacyl-ARNt synthétases[71]. Cet aminoacyl-ARNt peut alors être traité par un ribosome, dont la fonction est de lier les acides aminés entre eux en suivant la séquence indiquée par l'ARN messager transcrit à partir des gènes[72].
Voie de novo et voie de sauvetage des nucléotides |
Les nucléotides sont produits à partir d'acides aminés, de dioxyde de carbone et de formiate à travers des voies métaboliques qui consomment beaucoup d'énergie[73]. C'est la raison pour laquelle la plupart des organismes disposent de systèmes efficaces pour récupérer les nucléotides déjà existants[73],[74]. Les purines sont produites sous forme de nucléosides, c'est-à-dire d'une base nucléique liée au ribose. L'adénine et la guanine dérivent toutes deux de l'inosine monophosphate (IMP), produite à partir d'atomes issus de la glycine, de la glutamine, de l'aspartate et du formiate transféré au tétrahydrofolate. Les pyrimidines, quant à elles, sont produites à partir de l'orotate, lui-même issu de la glutamine et de l'aspartate[75].
Xénobiotiques et stress oxydant |
Tous les organismes sont exposés en permanence à des espèces chimiques qu'ils ne peuvent pas utiliser comme nutriments et qui pourraient être dangereuses si elles s'accumulaient dans les cellules, n'apportant aucun bénéfice métabolique. De tels composés sont appelés xénobiotiques[76]. L'organisme peut détoxiquer certains d'entre eux tels que les drogues, les poisons et les antibiotiques à l'aide de groupes d'enzymes spécifiques. Chez l'homme, de telles enzymes comprennent les cytochromes P450[77], les glucuronosyltransférases[78] et les glutathion S-transférases[79]. Ce système enzymatique agit en trois étapes pour d'abord oxyder le xénobiotique (phase I) puis conjuguer des groupes hydrosolubles sur le composé (phase II) et enfin pomper ce dernier hors des cellules pour être éventuellement encore métabolisé chez les organismes multicellulaires avant d'être finalement excrété (phase III). Ces réactions sont particulièrement importantes du point de vue écologique car elles interviennent dans la dégradation microbienne des polluants et la bioremédiation des sols contaminés et des marées noires[80]. De nombreuses réactions métaboliques microbiennes sont présentes également chez les organismes multicellulaires mais, compte tenu de l'extrême diversité des organismes unicellulaires, ces derniers sont en mesure de traiter un bien plus grand nombre de xénobiotiques que les multicellulaires et peuvent dégrader jusqu'aux polluants persistants tels que les composés organochlorés[81].
Les organismes aérobies sont confrontés au stress oxydant[82]. En effet, la phosphorylation oxydative et la formation des ponts disulfure indispensables au repliement de nombreuses protéines produisent des dérivés réactifs de l'oxygène tels que le peroxyde d'hydrogène[83]. Ces oxydants dangereux sont traités par des antioxydants tels que le glutathion et des enzymes telles que les catalases et les peroxydases[84],[85].
Régulation et contrôle du métabolisme |
Les êtres vivants étant soumis à de constants changements de leur environnement, leur métabolisme doit être continuellement adapté pour maintenir leurs constantes physiologiques — comme la température et la concentration intracellulaire des différentes espèces chimiques — dans un intervalle de valeurs normales, ce qu'on appelle l'homéostasie[86],[87]. La régulation du métabolisme permet également aux êtres vivants de répondre aux stimulus et d'interagir avec leur environnement[88]. Deux mécanismes apparentés sont particulièrement importants pour comprendre les modes de contrôle du métabolisme cellulaire : d'une part, la régulation d'une enzyme est la modulation de la cinétique réactionnelle de cette enzyme, c'est-à-dire l'accroissement ou la réduction de son activité en réponse à divers signaux chimiques, et, d'autre part, le contrôle exercé par une enzyme est l'effet de ses variations d'activité sur l'activité globale d'une voie métabolique, représentée par le flux de métabolites qui empruntent cette voie[89]. En effet, une enzyme peut être fortement régulée, et ainsi montrer d'importantes variations d'activité, tout en n'ayant pas d'incidence sur le flux global de métabolites à travers une voie dans laquelle elle intervient, de sorte qu'une telle enzyme n'exerce pas de contrôle sur cette voie métabolique[90].
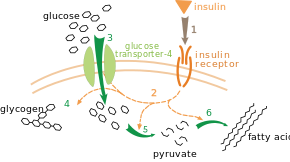
(en) Action de l'insuline sur l'absorption et le métabolisme du glucose. L'insuline se lie à son récepteur (1), ce qui déclenche une cascade d'activations de protéines et d'enzymes (2), notamment la translocation du transporteur de glucose de type 4 vers la membrane plasmique et l'entrée de glucose dans la cellule (3), la synthèse du glycogène (4), la glycolyse (5) et la biosynthèse des acides gras (6).
Il existe plusieurs niveaux de régulation du métabolisme. La régulation intrinsèque est l'autorégulation d'une voie métabolique en réponse aux changements de concentration des substrats ou des produits. Ainsi, la baisse de la concentration du produit d'une voie métabolique peut accroître le flux de métabolites à travers cette voie pour compenser la raréfaction de ce composé dans la cellule[89]. Ce type de régulation repose souvent sur la régulation allostérique de plusieurs enzymes de la voie métabolique[91]. Le contrôle extrinsèque concerne les cellules d'organismes multicellulaires répondant aux signaux d'autres cellules. Ces signaux prennent généralement la forme de « messagers hydrosolubles », tels que des hormones et les facteurs de croissance, qui sont détectés par des récepteurs membranaires spécifiques à la surface des cellules[92]. Ces signaux sont transmis à l'intérieur de la cellule par un mécanisme de transduction de signal faisant intervenir des messagers secondaires qui agissent souvent à travers la phosphorylation de certaines protéines[93].
Un exemple de contrôle extrinsèque très bien compris est la régulation du métabolisme du glucose par l'insuline[94]. L'insuline est produite en réponse à l'augmentation de la glycémie, c'est-à-dire du taux de glucose dans le sang. La liaison de cette hormone à ses récepteurs cellulaires active une cascade de protéine kinases qui conduisent les cellules à absorber du glucose et à le convertir en molécules de stockage telles que des acides gras et du glycogène[95]. Le métabolisme du glycogène est contrôlé par l'activité de la glycogène phosphorylase, qui dégrade le glycogène, et de la glycogène synthase, qui le produit. Ces enzymes font l'objet d'une régulation symétrique, la phosphorylation activant la glycogène phosphorylase mais inhibant la glycogène synthase. L'insuline favorise la production de glycogène en activant des phosphatases qui réactivent la glycogène synthase et désactivent la glycogène phosphorylase en réduisant leur phosphorylation[96].
Évolution |

Arbre phylogénétique montrant les filiations génétiques des êtres vivants dans les trois domaines du vivant : les bactéries figurent en bleu, les eucaryotes en rouge et les archées en vert. Certains embranchements sont positionnés sur l'arbre.
Les grandes voies métaboliques évoquées plus haut, telles que la glycolyse et le cycle de Krebs, sont présentes chez les organismes appartenant aux trois domaines du vivant : bactéries, eucaryotes et archées. Il est possible qu'elles remontent toutes les trois à un dernier ancêtre commun universel[3],[97], vraisemblablement procaryotique et peut-être méthanogène doté d'un métabolisme complet des acides aminés, des nucléotides, des glucides et des lipides[98],[99] ; les chlorobactéries pourraient être les plus anciens organismes encore vivants[100]. La conservation au cours de l'évolution de ces voies métaboliques anciennes pourrait provenir du fait qu'elles sont apparues comme des solutions optimales à des problèmes métaboliques particuliers, la glycolyse et le cycle de Krebs produisant leurs métabolites de façon efficace et en un minimum d'étapes[4],[5]. Il est possible que les premières voies métaboliques fondées sur des enzymes aient été relatives aux purines, tandis que les voies alors préexistantes seraient apparues dans un monde à ARN fondé sur des ribozymes[101].
De nombreux modèles ont été proposés pour décrire les mécanismes par lesquels de nouvelles voies métaboliques apparaissent. Cela passe par l'addition séquentielle de nouvelles enzymes à des voies plus courtes, la duplication ou la divergence de voies préexistantes ou encore l'intégration d'enzymes préexistantes dans des voies métaboliques nouvelles[102]. L'importance relative de ces différents mécanismes reste obscure, mais la génomique a montré que les enzymes d'une même voie métabolique ont de fortes chances de partager un ancêtre commun, ce qui tendrait à montrer que de nombreuses voies ont évolué progressivement par apparition de nouvelles fonctionnalités à partir d'étapes préexistantes dans la voie métabolique en question[103]. Un autre modèle provenant d'études sur l'évolution des structures protéiques impliquées dans les réseaux de voies métaboliques a suggéré que les enzymes y sont très largement intégrées pour réaliser des fonctions semblables dans différentes voies métaboliques, ce qui apparaît clairement dans la base de données MANET[104]. Ces processus d'intégration se déroulent selon un modèle en mosaïque[105]. Une troisième possibilité est la présence de certains segments de voies métaboliques utilisables de façon modulaire pour faire apparaître d'autres voies métaboliques et réaliser des fonctions semblables sur des molécules différentes[106].
Outre l'apparition de nouvelles voies métaboliques, l'évolution peut également faire disparaître certaines fonctionnalités biochimiques. C'est par exemple le cas chez certains parasites, qui tendent à absorber les biomolécules de leur hôte et à perdre la capacité à les synthétiser eux-mêmes[107]. On observe une semblable réduction des aptitudes métaboliques chez les organismes endosymbiotiques[108].
Thermodynamique du métabolisme |
Le métabolisme est soumis aux principes de la thermodynamique, qui régissent les échanges de chaleur et de travail. Le deuxième principe de la thermodynamique indique que, dans tout système fermé, l'entropie (c'est-à-dire le désordre) tend à augmenter. Bien que l'extrême complexité des êtres vivants semble en contradiction avec ce principe, la vie n'est cependant possible que parce que tous les organismes sont des systèmes ouverts, qui échangent matière et énergie avec leur environnement. Par conséquent, les êtres vivants ne sont pas en équilibre, mais sont des systèmes dissipatifs qui maintiennent leur haut degré de complexité par l'augmentation plus importante de l'entropie de leur environnement[109]. Le métabolisme cellulaire y parvient en couplant les processus spontanés du catabolisme avec les processus non spontanés de l'anabolisme : du point de vue thermodynamique, le métabolisme maintient l'ordre en créant le désordre[110].
Exemples de réactions métaboliques |

Graphique énergétique.
Le métabolisme de dégradation de grosses molécules en petites molécules, qui permet la libération d'énergie, est appelé catabolisme. L'énergie est mise en réserve lors de la phosphorylation de l'ADP (adénosine diphosphate) en ATP (adénosine-triphosphate). Cette énergie servira à assurer les différentes fonctions de la cellule.
Trois modes de productions principaux d’énergie :
métabolisme anaérobie alactique : il fournit une grande quantité d’énergie sur une courte durée, par dégradation des faibles réserves d'ATP en donnant de l'ADP ;
métabolisme anaérobie lactique : l'ATP est créé sans dioxygène au prix d'une fermentation lactique donnant un poison cellulaire, l'acide lactique ;
métabolisme aérobie : avec un apport en dioxygène normal, on observe une respiration cellulaire classique.
Cependant diverses voies métaboliques existent comme en témoigne cette image :
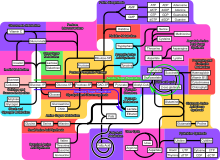
Voies métaboliques.
Métabolisme et température |
Chez les organismes fongiques, bactériens, végétaux ou animaux à sang chaud ou froid, divers processus font interagir la température interne, externe et le métabolisme[111], avec des boucles de rétroactions plus ou moins complexes, variant selon les espèces, les individus, leur forme et taille[111] et leur masse corporelle[112] et les milieux[113].
Plantes et levures semblent disposer d'un thermostat biologique simple ; Chez l’arabette Arabidopsis thaliana, une seule protéine (l'histone H2A.Z) joue ce rôle pour des variations de température de moins de 1 °C. Cette protéine modifie l’enroulement de l’ADN sur lui-même et contrôle ainsi l’accès à l’ADN de certaines molécules inhibant ou activant plusieurs dizaines de gènes. Cet effet « bio-thermostat » semble fréquent dans la nature, car également détecté chez des organismes aussi différents que la levure et une crucifère commune[114],[115].
La compréhension de ces mécanismes devrait aussi aider à mieux comprendre certains effets (sur les gènes) du dérèglement climatique.
Métabonomique |
La métabonomique mesure l'empreinte des perturbations biochimiques causées par les maladies, les médicaments ou des produits toxiques[116]. Introduite dans les années 1980, cette discipline n'a commencé à jouer un rôle important en recherche et développement dans l’industrie pharmaceutique qu'au XXIe siècle. Complémentaire de la génomique et de la protéomique, elle permet par exemple de caractériser les modèles animaux de diverses pathologies afin d’identifier de nouvelles cibles pharmacologiques. La particularité de la métabonomique est l'analyse simultanée d'un très grand nombre de métabolites, c'est-à-dire de petites molécules intermédiaires des voies métaboliques, dans les milieux biologiques tels que l'urine ou le plasma. Des outils de screening (exploration large et systématique) métabolique tels que la résonance magnétique nucléaire ou la spectrométrie de masse sont utilisés afin d’identifier des marqueurs de toxicité (ou des séries de marqueurs, correspondant à des profils métaboliques), dans le but de déceler, tôt dans le cycle de développement, les médicaments candidats qui présenteront des effets indésirables. Idéalement, les biomarqueurs identifiés en phase préclinique seront non-invasifs et utilisables en phase clinique pour suivre le déclenchement, la progression et la guérison d’une pathologie. Afin d’identifier de nouveaux métabolites marqueurs de toxicité, il est également nécessaire de connaître les variations dites « normales » du pool métabolique (effet du rythme circadien, du stress, du régime alimentaire, de l'amaigrissement, etc.). Il est ainsi possible de découvrir les perturbations métaboliques qui sont spécifiques de la pathologie étudiée.
Autre contenu sémantique |
Métaphoriquement et par extension on parle parfois de métabolisme urbain, industriel, social[117],[118] ou sociétal[119] pour décrire les intrants (ressources naturelles, foncières, humaines...) et extrants (déchets) qui caractérisent ces systèmes.
Notes et références |
(en) Cornelius G. Friedrich, « Physiology and Genetics of Sulfur-oxidizing Bacteria », Advances in Microbial Physiology (en), vol. 39, 1997, p. 235-289 (PMID 9328649, DOI 10.1016/S0065-2911(08)60018-1, lire en ligne)
(en) Norman R. Pace, « The universal nature of biochemistry », Proceedings of the National Academy of Sciences of the United States of America, vol. 98, no 3, 30 janvier 2001, p. 805-808 (PMID 11158550, DOI 10.1073/pnas.98.3.805, lire en ligne)
(en) Eric Smith et Harold J. Morowitz, « Universality in intermediary metabolism », Proceedings of the National Academy of Sciences of the United States of America, vol. 101, no 36, 7 septembre 2004, p. 13168-13173 (PMID 15340153, DOI 10.1073/pnas.0404922101, lire en ligne)
(en) Oliver Ebenhöh et Reinhart Heinrich, « Evolutionary optimization of metabolic pathways. Theoretical reconstruction of the stoichiometry of ATP and NADH producing systems », Bulletin of Mathematical Biology, vol. 63, no 1, janvier 2001, p. 21-55 (PMID 11146883, DOI 10.1006/bulm.2000.0197, lire en ligne)
(en) Enrique Meléndez-Hevia, Thomas G. Waddell et Marta Cascante, « The puzzle of the Krebs citric acid cycle: Assembling the pieces of chemically feasible reactions, and opportunism in the design of metabolic pathways during evolution », Journal of Molecular Evolution, vol. 43, no 3, septembre 1996, p. 293-303 (PMID 8703096, DOI 10.1007/BF02338838, lire en ligne)
(en) Katharine A. Michie et Jan Löwe, « Dynamic Filaments of the Bacterial Cytoskeleton », Annual Review of Biochemistry, vol. 75, juillet 2006, p. 467-492 (lire en ligne) DOI:10.1146/annurev.biochem.75.103004.142452
(en) A. L. McCall, W. R. Millington et R. J. Wurtman, « Metabolic fuel and amino acid transport into the brain in experimental diabetes mellitus », Proceedings of the National Academy of Sciences of the United States of America, vol. 79, no 17, septembre 1982, p. 5406-5410 (PMID 6752947, PMCID 346906, lire en ligne)
(en) John S. Hothersall et Aamir Ahmed, « Metabolic Fate of the Increased Yeast Amino Acid Uptake Subsequent to Catabolite Derepression », Journal of Amino Acids, vol. 2013, 2013, e461901 (PMCID 3575661, lire en ligne) DOI:10.1155/2013/461901
(en) Eoin Fahy, Shankar Subramaniam, H. Alex Brown, Christopher K. Glass, Alfred H. Merrill Jr., Robert C. Murphy, Christian R. H. Raetz, David W. Russell, Yousuke Seyama, Walter Shaw, Takao Shimizu, Friedrich Spener, Gerrit van Meer, Michael S. VanNieuwenhze, Stephen H. White, Joseph L. Witztum et Edward A. Dennis, « A comprehensive classification system for lipids », Journal of Lipid Research, vol. 46, mai 2005, p. 839-862 (PMID 15722563, DOI 10.1194/jlr.E400004-JLR200, lire en ligne)
(en) Fausto G. HEGARDT, « Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis », Biochemical Journal, vol. 338, 1999, p. 569-582 (PMID 10051425, PMCID 1220089, DOI 10.1042/0264-6021:3380569, lire en ligne)
(en) Rahul Raman, S. Raguram, Ganesh Venkataraman, James C. Paulson et Ram Sasisekharan, « Glycomics: an integrated systems approach to structure-function relationships of glycans », Nature Methods, vol. 2, 2005, p. 817-824 (DOI 10.1038/nmeth807, lire en ligne)
(en) Saleta Sierra, Bernd Kupfer et Rolf Kaiser, « Basics of the virology of HIV-1 and its replication », Journal of Clinical Virology, vol. 34, no 4, décembre 2005, p. 233-244 (PMID 16198625, DOI 10.1016/j.jcv.2005.09.004, lire en ligne)
(en) Peter MITCHELL, « Compartmentation and Communication in Living Systems. Ligand Conduction: a General Catalytic Principle in Chemical, Osmotic and Chemiosmotic Reaction Systems », European Journal of Biochemistry, vol. 95, no 1, mars 1979, p. 1-20 (PMID 378655, DOI 10.1111/j.1432-1033.1979.tb12934.x, lire en ligne)
(en) M. J. Wimmer et I. A. Rose, « Mechanisms of Enzyme-Catalyzed Group Transfer Reactions », Annual Review of Biochemistry, vol. 47, juillet 1978, p. 1031-1078 (PMID 354490, DOI 10.1146/annurev.bi.47.070178.005123, lire en ligne)
(en) Peter Dimroth, Christoph von Ballmoos et T. Meier, « Catalytic and mechanical cycles in F-ATP synthases », EMBO reports, vol. 7, 2006, p. 276-282 (PMID 16607397, PMCID 1456893, DOI 10.1038/sj.embor.7400646, lire en ligne)
(en) Nadine POLLAK, Christian DÖLLE et Mathias ZIEGLER, « The power to reduce: pyridine nucleotides – small molecules with a multitude of functions », Biochemical Journal, vol. 402, 2007, p. 205-218 (PMID 17295611, PMCID 1798440, DOI 10.1042/BJ20061638, lire en ligne)
(en) S. B. Heymsfield, M. Waki, J. Kehayias, S. Lichtman, F. A. Dilmanian, Y. Kamen, J. Wang et R. N. Pierson Jr, « Chemical and elemental analysis of humans in vivo using improved body composition models », American Journal of Physiology - Endocrinology and Metabolism, vol. 261, août 1991, E190-E198 (PMID 1872381, lire en ligne)
(en) A.F. Dulhunty, « EXCITATION–CONTRACTION COUPLING FROM THE 1950s INTO THE NEW MILLENNIUM », Clinical and Experimental Pharmacology and Physiology, vol. 33, no 9, septembre 2006, p. 763-772 (PMID 16922804, DOI 10.1111/j.1440-1681.2006.04441.x, lire en ligne)
(en) D. C. Mahan et R. G. Shields Jr, « Macro- and micromineral composition of pigs from birth to 145 kilograms of body weight », Journal of Animal Science, vol. 76, no 2, février 1998, p. 506-512 (PMID 9498359, lire en ligne)
(en) Søren Husted, Birgitte F. Mikkelsen, Jacob Jensen et Niels Erik Nielsen, « Elemental fingerprint analysis of barley (Hordeum vulgare) using inductively coupled plasma mass spectrometry, isotope-ratio mass spectrometry, and multivariate statistics », Analytical and Bioanalytical Chemistry, vol. 378, no 1, 2004, p. 171-182 (PMID 14551660, DOI 10.1007/s00216-003-2219-0, lire en ligne)
(en) Lydia A. Finney, Thomas V. O'Halloran, « Transition Metal Speciation in the Cell: Insights from the Chemistry of Metal Ion Receptors », Science, vol. 300, no 5621, 9 mai 2003, p. 931-936 (PMID 12738850, DOI 10.1126/science.1085049, lire en ligne)
(en) Robert J. Cousins, Juan P. Liuzzi et Louis A. Lichten, « Mammalian Zinc Transport, Trafficking, and Signals », Journal of Biological Chemistry, vol. 281, 25 août 2006, p. 24085-24089 (PMID 16793761, DOI 10.1074/jbc.R600011200, lire en ligne)
(en) Louise L. Dunn, Yohan Suryo Rahmanto et Des R. Richardson, « Iron uptake and metabolism in the new millennium », Trends in Cell Biology, vol. 17, no 2, février 2007, p. 93-100 (PMID 17194590, DOI 10.1016/j.tcb.2006.12.003, lire en ligne)
(en) Kenneth H. Nealson et Pamela G. Conrad, « Life: past, present and future », Philosophical Transactions of the Royal Society B Biological Sciences, vol. 354, no 1392, 29 décembre 1999, p. 1923-1939 (PMID 10670014, PMCID 1692713, DOI 10.1098/rstb.1999.0532, lire en ligne)
(en) C. C. Häse et R. A. Finkelstein, « Bacterial extracellular zinc-containing metalloproteases », Microbiology and Molecular Biology Reviews, vol. 57, no 4, décembre 1993, p. 823-837 (PMID 8302217, PMCID 372940, lire en ligne)
(en) R. Gupta, N. Gupta et P. Rathi, « Bacterial lipases: an overview of production, purification and biochemical properties », Applied Microbiology and Biotechnology, vol. 64, no 6, juin 2004, p. 763-781 (PMID 14966663, DOI 10.1007/s00253-004-1568-8, lire en ligne)
(en) Wiley W. Souba et Anthony J. Pacitti, « Review: How Amino Acids Get Into Cells: Mechanisms, Models, Menus, and Mediators », Journal of Parental & Enteral Nutrition, vol. 16, no 6, novembre 1992, p. 569-578 (PMID 1494216, DOI 10.1177/0148607192016006569, lire en ligne)
(en) Michael P Barrett, Adrian R Walmsleyt et Gwyn W Gould, « Structure and function of facultative sugar transporters », Current Opinion in Cell Biology, vol. 11, no 4, août 1999, p. 496–502 (PMID 10449337, DOI 10.1016/S0955-0674(99)80072-6, lire en ligne)
(en) G. I. Bell, C. F. Burant, J. Takeda et G. W. Gould, « Structure and function of mammalian facilitative sugar transporters », Journal of Biological Chemistry, vol. 268, 1993, p. 19161-19164 (PMID 8366068, lire en ligne)
(en) Clara Bouché, Shanti Serdy, C. Ronald Kahn et Allison B. Goldfine, « The Cellular Fate of Glucose and Its Relevance in Type 2 Diabetes », Endocrine Reviews, vol. 25, no 5, octobre 2004, p. 807-830 (PMID 15466941, DOI 10.1210/er.2003-0026, lire en ligne)
(en) W. Sakami et H. Harrington, « Amino Acid Metabolism », Annual Reviews, vol. 32, juillet 1963, p. 355-398 (PMID 14144484, DOI 10.1146/annurev.bi.32.070163.002035, lire en ligne)
(en) John T. Brosnan, « Glutamate, at the Interface between Amino Acid and Carbohydrate Metabolism », Journal of Nutrition, vol. 130, no 4, avril 2000, p. 988S-990S (PMID 10736367, lire en ligne)
(en) Vernon R. Young et Alfred M. Ajami, « Glutamine: The Emperor or His Clothes? », Journal of Nutrition, vol. 131, no 9, septembre 2001, p. 2449S-2459S (PMID 11533293, lire en ligne)
(en) Jonathan P. Hosler, Shelagh Ferguson-Miller et Denise A. Mills, « Energy Transduction: Proton Transfer Through the Respiratory Complexes », Annual Review of Biochemistry, vol. 75, juillet 2006, p. 165-187 (DOI 10.1146/annurev.biochem.75.062003.101730, lire en ligne)
(en) Brian E. Schultz et Sunney I. Chan, « STRUCTURES AND PROTON-PUMPING STRATEGIES OF MITOCHONDRIAL RESPIRATORY ENZYMES », Annual Review of Biophysics and Biomolecular Structure, vol. 30, juin 2001, p. 23-65 (PMID 11340051, DOI 10.1146/annurev.biophys.30.1.23, lire en ligne)
(en) Roderick A Capaldi et Robert Aggeler, « Mechanism of the F1FO-type ATP synthase, a biological rotary motor », Trends in Biochemical Sciences, vol. 27, no 3, mars 2002, p. 154-160 (PMID 11893513, DOI 10.1016/S0968-0004(01)02051-5, lire en ligne)
(en) B. Friedrich et E. Schwartz, « Molecular Biology of Hydrogen Utilization in Aerobic Chemolithotrophs », Annual Review of Microbiology, vol. 47, octobre 1993, p. 351-383 (PMID 8257102, DOI 10.1146/annurev.mi.47.100193.002031, lire en ligne)
(en) Karrie A. Weber, Laurie A. Achenbach et John D. Coates, « Microorganisms pumping iron: anaerobic microbial iron oxidation and reduction », Nature Reviews Microbiology, vol. 4, octobre 2006, p. 752-764 (PMID 16980937, DOI 10.1038/nrmicro1490, lire en ligne)
(en) Mike S.M Jetten, Marc Strous, Katinka T van de Pas-Schoonen, Jos Schalk, Udo G.J.M van Dongen, Astrid A van de Graaf, Susanne Logemann, Gerard Muyzer, Mark C.M van Loosdrecht et J.Gijs Kuenen, « The anaerobic oxidation of ammonium », FEMS Microbiology Reviews, vol. 22, no 5, décembre 1998, p. 421-437 (PMID 9990725, DOI 10.1111/j.1574-6976.1998.tb00379.x, lire en ligne)
(en) Jörg Simon, « Enzymology and bioenergetics of respiratory nitrite ammonification », FEMS Microbiology Reviews, vol. 26, no 3, août 2002, p. 285-309 (PMID 12165429, DOI 10.1111/j.1574-6976.2002.tb00616.x, lire en ligne)
(en) R. Conrad, « Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO) », Microbiology and Molecular Biology Reviews, vol. 60, no 4, décembre 1996, p. 609-640 (PMID 8987358, PMCID 239458, lire en ligne)
(en) José-Miguel Barea, María José Pozo, Rosario Azcón et Concepción Azcón-Aguilar, « Microbial co-operation in the rhizosphere », Journal of Experimental Botany, vol. 56, no 417, juillet 2005, p. 1761-1778 (DOI 10.1093/jxb/eri197, lire en ligne)
(en) Marcel T. J. van der Meer, Stefan Schouten, Mary M. Bateson, Ulrich Nübel, Andrea Wieland, Michael Kühl, Jan W. de Leeuw, Jaap S. Sinninghe Damsté et David M. Ward, « Diel Variations in Carbon Metabolism by Green Nonsulfur-Like Bacteria in Alkaline Siliceous Hot Spring Microbial Mats from Yellowstone National Park », Applied and Environmental Microbiology, vol. 71, no 7, juillet 2005, p. 3978-3986 (PMID 16000812, PMCID 1168979, DOI 10.1128/AEM.71.7.3978-3986.2005, lire en ligne)
(en) Mary A. Tichi et F. Robert Tabita, « Interactive Control of Rhodobactercapsulatus Redox-Balancing Systems during Phototrophic Metabolism », Applied and Environmental Microbiology, vol. 183, no 21, novembre 2001, p. 6344-6354 (PMID 11591679, PMCID 100130, DOI 10.1128/JB.183.21.6344-6354.2001, lire en ligne)
(en) J.P. Allen et J.C. Williams, « Photosynthetic reaction centers », FEBS Letters, vol. 438, nos 1-2, 30 octobre 1998, p. 5-9 (DOI 10.1016/S0014-5793(98)01245-9, lire en ligne)
(en) Yuri Munekage, Mihoko Hashimoto, Chikahiro Miyake, Ken-Ichi Tomizawa, Tsuyoshi Endo, Masao Tasaka et Toshiharu Shikanai, « Cyclic electron flow around photosystem I is essential for photosynthesis », Nature, vol. 429, 3 juin 2004, p. 579-582 (DOI 10.1038/nature02598, lire en ligne)
(en) H. M. Miziorko et G. H. Lorimer, « Ribulose-1,5-Bisphosphate Carboxylase-Oxygenase », Annual Review of Biochemistry, vol. 52, juillet 1983, p. 507-535 (PMID 6351728, DOI 10.1146/annurev.bi.52.070183.002451, lire en ligne)
(en) Antony N. Dodd, Anne M. Borland, Richard P. Haslam, Howard Griffiths et Kate Maxwell, « Crassulacean acid metabolism: plastic, fantastic », Journal of Experimental Botany, vol. 53, no 369, 2002, p. 569-580 (PMID 11886877, DOI 10.1093/jexbot/53.369.569, lire en ligne)
(en) Michael Hügler, Carl O. Wirsen, Georg Fuchs, Craig D. Taylor et Stefan M. Sievert, « Evidence for Autotrophic CO2 Fixation via the Reductive Tricarboxylic Acid Cycle by Members of the ε Subdivision of Proteobacteria », Journal of Bacteriology, vol. 187, no 9, mai 2005, p. 3020-3027 (PMID 15838028, PMCID 1082812, DOI 10.1128/JB.187.9.3020-3027.2005, lire en ligne)
(en) Gerhard STRAUSS et Georg FUCHS, « Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3-hydroxypropionate cycle », European Journal of Biochemistry, vol. 215, no 3, août 1993, p. 633-643 (PMID 8354269, DOI 10.1111/j.1432-1033.1993.tb18074.x, lire en ligne)
(en) H. G. Wood, « Life with CO or CO2 and H2 as a source of carbon and energy », FASEB Journal, vol. 5, no 2, février 1991, p. 156-163 (PMID 1900793, lire en ligne)
(en) Jessup M. Shively, Geertje van Keulen, et Wim G. Meijer, « SOMETHING FROM ALMOST NOTHING: Carbon Dioxide Fixation in Chemoautotrophs », Annual Review of Microbiology, vol. 52, octobre 1998, p. 191-230 (PMID 9891798, DOI 10.1146/annurev.micro.52.1.191, lire en ligne)
(en) A. Boiteux et B. Hess, « Design of Glycolysis », Philosophical Transactions of the Royal Society B Biological Sciences, vol. 293, no 1063, 26 juin 1981, p. 5-22 (PMID 6115423, DOI 10.1098/rstb.1981.0056, lire en ligne)
(en) S. J. Pilkis, M. R. El-Maghrabi et T. H. Claus, « Fructose-2,6-Bisphosphate in Control of Hepatic Gluconeogenesis: From Metabolites to Molecular Genetics », Diabetes Care, vol. 13, no 6, juin 1990, p. 582-599 (PMID 2162755, DOI 10.2337/diacare.13.6.582, lire en ligne)
(en) Scott A. Ensign, « Revisiting the glyoxylate cycle: alternate pathways for microbial acetate assimilation », Molecular Microbiology, vol. 61, no 2, juillet 2006, p. 274-276 (PMID 16856935, DOI 10.1111/j.1365-2958.2006.05247.x, lire en ligne)
(en) Patrick F. Finn et J. Fred Dice, « Proteolytic and lipolytic responses to starvation », Nutrition, vol. 22, no 7, juillet 2006, p. 830-844 (PMID 16815497, DOI 10.1016/j.nut.2006.04.008, lire en ligne)
(en) H. L. KORNBERG et H. A. KREBS, « Synthesis of Cell Constituents from C2-Units by a Modified Tricarboxylic Acid Cycle », Nature, vol. 179, 18 mai 1957, p. 988-991 (PMID 13430766, DOI 10.1038/179988a0, Bibcode 1957Natur.179..988K, lire en ligne)
(en) T. W. Rademacher, R. B. Parekh et R. A. Dwek, « Glycobiology », Annual Review of Biochemistry, vol. 57, juillet 1988, p. 785-838 (PMID 3052290, DOI 10.1146/annurev.bi.57.070188.004033, lire en ligne)
(en) G. Opdenakker, P. M. Rudd, C. P. Ponting et R. A. Dwek, « Concepts and principles of glycobiology », FASEB Journal, vol. 7, no 14, novembre 1993, p. 1330-1337 (PMID 8224606, lire en ligne)
(en) Malcolm J. McConville et Anant K. Menon, « Recent developments in the cell biology and biochemistry of glycosylphosphatidylinositol lipids (Review) », Molecular Membrane Biology, vol. 17, no 1, 2000, p. 1-16 (PMID 10824734, DOI 10.1080/096876800294443, lire en ligne)
(en) Subrahmanyam S. Chirala et Salih J. Wakil, « Structure and function of animal fatty acid synthase », Lipids, vol. 39, no 11, novembre 2004, p. 1045-1053 (PMID 15726818, DOI 10.1007/s11745-004-1329-9, lire en ligne)
(en) Stephen W. White, Jie Zheng, Yong-Mei Zhang et Charles O. Rock, « THE STRUCTURAL BIOLOGY OF TYPE II FATTY ACID BIOSYNTHESIS », Annual Review of Biochemistry, vol. 74, juillet 2005, p. 791-831 (PMID 15952903, DOI 10.1146/annurev.biochem.74.082803.133524, lire en ligne)
(en) John B. Ohlrogge et Jan G. Jaworski, « REGULATION OF FATTY ACID SYNTHESIS », Annual Review of Plant Physiology and Plant Molecular Biology, vol. 48, juin 1997, p. 109-136 (PMID 15012259, DOI 10.1146/annurev.arplant.48.1.109, lire en ligne)
(en) Vinod Shanker Dubey, Ritu Bhalla et Rajesh Luthra, « An overview of the non-mevalonate pathway for terpenoid biosynthesis in plants », Journal of Biosciences, vol. 28, no 5, septembre 2003, p. 637-646 (PMID 14517367, DOI 10.1007/BF02703339, lire en ligne)
(en) Tomohisa Kuzuyama et Haruo Seto, « Diversity of the biosynthesis of the isoprene units », Natural Product Reports, vol. 20, no 2, 2003, p. 171-183 (PMID 12735695, DOI 10.1039/B109860H, lire en ligne)
(en) Laura L. Grochowski, Huimin Xu et Robert H. White, « Methanocaldococcus jannaschii Uses a Modified Mevalonate Pathway for Biosynthesis of Isopentenyl Diphosphate », Journal of Bacteriology, vol. 188, no 9, mai 2006, p. 3192-3198 (PMID 16621811, PMCID 1447442, DOI 10.1128/JB.188.9.3192-3198.2006, lire en ligne)
(en) Hartmut K. Lichtenthaler, « THE 1-DEOXY-D-XYLULOSE-5-PHOSPHATE PATHWAY OF ISOPRENOID BIOSYNTHESIS IN PLANTS », Annual Review of Plant Physiology and Plant Molecular Biology, vol. 50, juin 1999, p. 47-65 (PMID 15012203, DOI 10.1146/annurev.arplant.50.1.47, lire en ligne)
(en) G J Schroepfer, Jr, « Sterol Biosynthesis », Annual Review of Biochemistry, vol. 50, juillet 1981, p. 585-621 (PMID 7023367, DOI 10.1146/annurev.bi.50.070181.003101, lire en ligne)
(en) N. D. Lees, B. Skaggs, D. R. Kirsch et M. Bard, « Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae—A review », Lipids, vol. 30, no 3, mars 1995, p. 221-226 (PMID 7791529, DOI 10.1007/BF02537824, lire en ligne)
(en) Ralf Himmelreich, Helmut Hilbert, Helga Plagens, Elsbeth Pirkl, Bi-Chen Li et Richard Herrmann, « Complete Sequence Analysis of the Genome of the Bacterium Mycoplasma pneumoniae », Nucleic Acids Research, vol. 24, no 22, 1996, p. 4420-4449 (PMID 8948633, PMCID 146264, DOI 10.1093/nar/24.22.4420, lire en ligne)
(en) Michael Ibba et Dieter Söll, « The renaissance of aminoacyl-tRNA synthesis », EMBO Reports, vol. 2, no 5, mai 2001, p. 382-387 (PMID 11375928, PMCID 1083889, DOI 10.1093/embo-reports/kve095, lire en ligne)
(en) PD Lengyel et D. Söll, « Mechanism of protein biosynthesis », Bacteriological Reviews, juin 1969, p. 264-301 (PMID 4896351, PMCID 378322, lire en ligne)
(en) Rita Zrenner, Mark Stitt, Uwe Sonnewald et Ralf Boldt, « PYRIMIDINE AND PURINE BIOSYNTHESIS AND DEGRADATION IN PLANTS », Annual Review of Plant Biology, vol. 57, juin 2006, p. 805-836 (PMID 16669783, DOI 10.1146/annurev.arplant.57.032905.105421, lire en ligne)
(en) Claudio Stasolla, Riko Katahira, Trevor A. Thorpe et Hiroshi Ashihara, « Purine and pyrimidine nucleotide metabolism in higher plants », Journal of Plant Physiology, vol. 160, no 11, 2003, p. 1271-1295 (PMID 14658380, DOI 10.1078/0176-1617-01169, lire en ligne)
(en) Janet L Smith, « Enzymes of nucleotide synthesis », Current Opinion in Structural Biology, vol. 5, no 6, décembre 1995, p. 752-757 (PMID 8749362, DOI 10.1016/0959-440X(95)80007-7, lire en ligne)
(en) Bernard Testa et StefanieD. Krämer, « The Biochemistry of Drug Metabolism – An Introduction », Chemistry & Biodiversity, vol. 3, no 10, octobre 2006, p. 1053-1101 (PMID 17193224, DOI 10.1002/cbdv.200690111, lire en ligne)
(en) P.B. Danielson, « The Cytochrome P450 Superfamily: Biochemistry, Evolution and Drug Metabolism in Humans », Current Drug Metabolism, vol. 3, no 6, 2002, p. 561-597 (PMID 17193224, DOI 10.2174/1389200023337054, lire en ligne)
(en) C. D. King, G. R. Rios, M. D. Green et T. R. Tephly, « UDP-Glucuronosyltransferases », Current Drug Metabolism, vol. 1, no 2, 2000, p. 143–161 (PMID 11465080, DOI 10.2174/1389200003339171, lire en ligne)
(en) David SHEEHAN, Gerardene MEADE, Vivienne M. FOLEY et Catriona A. DOWD, « Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily », Biochemical Journal, vol. 360, 2001, p. 1-16 (PMID 11695986, PMCID 1222196, DOI 10.1042/0264-6021:3600001, lire en ligne)
(en) Teca Calcagno Galvão, William W. Mohn et Víctor de Lorenzo, « Exploring the microbial biodegradation and biotransformation gene pool », Trends in Biotechnology, vol. 23, no 10, octobre 2005, p. 497-506 (PMID 16125262, DOI 10.1016/j.tibtech.2005.08.002, lire en ligne)
(en) Dick B. Janssen, Inez J. T. Dinkla, Gerrit J. Poelarends et Peter Terpstra, « Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities », Environmental Microbiology, vol. 7, no 12, décembre 2005, p. 1868-1882 (PMID 16309386, DOI 10.1111/j.1462-2920.2005.00966.x, lire en ligne)
(en) Kelvin J. A. Davies, « Oxidative stress: the paradox of aerobic life », Biochemical Society Symposia, vol. 61, 1995, p. 1-31 (PMID 8660387, lire en ligne)
(en) Benjamin P. Tu et Jonathan S. Weissman, « Oxidative protein folding in eukaryotes – mechanisms and consequences », Journal of Cell Biology, vol. 164, no 3, 2 février 2004, p. 341-346 (PMID 14757749, PMCID 2172237, DOI 10.1083/jcb.200311055, lire en ligne)
(en) Helmut Sies, « Oxydative Stress: Oxidants and Antioxidants », Experimental Physiology, vol. 82, 1997, p. 291-295 (PMID 9129943, lire en ligne)
(en) Silvia Vertuani, Angela Angusti et Stefano Manfredini, « The Antioxidants and Pro-Antioxidants Network: An Overview », Current Pharmaceutical Design, vol. 10, no 14, 2004, p. 1677-1694 (PMID 15134565, DOI 10.2174/1381612043384655, lire en ligne)
(en) Réka Albert, « Scale-free networks in cell biology », Journal of Cell Science, vol. 118, novembre 2005, p. 4947-4957 (PMID 16254242, DOI 10.1242/jcs.02714, lire en ligne)
(en) M. D. Brand, « Regulation analysis of energy metabolism », Journal of Experimental Biology, vol. 200, janvier 1997, p. 193-202 (PMID 9050227, lire en ligne)
(en) Orkun S. Soyer, Marcel Salathé et Sebastian Bonhoeffer, « Signal transduction networks: Topology, response and biochemical processes », Journal of Theoretical Biology, vol. 238, no 2, 21 janvier 2006, p. 416-425 (PMID 16045939, DOI 10.1016/j.jtbi.2005.05.030, lire en ligne)
(en) M. Salter, R. G. Knowles, C. I. Pogson, « Metabolic control », Essays in Biochemistry, vol. 28, 1994, p. 1-12 (PMID 7925313)
(en) Hans V. WESTERHOFF, Albert K. GROEN et Ronald J. WANDERS, « Modern theories of metabolic control and their applications », Bioscience Reports, vol. 4, 1984, p. 1-22 (PMID 6365197, DOI 10.1007/BF01120819, lire en ligne)
(en) D. A. Fell et S. Thomas, « Physiological control of metabolic flux: the requirement for multisite modulation », Biochemical Journal, vol. 311, 1995, p. 35-39 (PMID 7575476, PMCID 1136115, lire en ligne)
(en) Wayne A. Hendrickson, « Transduction of biochemical signals across cell membranes », Quarterly Reviews of Biophysics, vol. 38, no 04, novembre 2005, p. 321-330 (PMID 16600054, DOI 10.1017/S0033583506004136, lire en ligne)
(en) Philip Cohen, « The regulation of protein function by multisite phosphorylation – a 25 year update », Trends in Biochemical Sciences, vol. 25, no 12, décembre 2000, p. 596-601 (PMID 11116185, DOI 10.1016/S0968-0004(00)01712-6, lire en ligne)
(en) Gustav E. Lienhard, Jan W. Slot, David E. James et Mike M. Mueckler, « How Cells Absorb Glucose », Scientific American, vol. 266, 1992, p. 86-91 (PMID 1734513, DOI 10.1038/scientificamerican0192-86, lire en ligne)
(en) Peter J. Roach, « Glycogen and its Metabolism », Current Molecular Medicine, vol. 2, no 2, 2002, p. 101-120 (PMID 11949930, DOI 10.2174/1566524024605761, lire en ligne)
(en) C. B. Newgard, M. J. Brady, R. M. O'Doherty et A. R. Saltiel, « Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1 », Diabetes, vol. 49, no 12, décembre 2000, p. 1967-1977 (PMID 11117996, DOI 10.2337/diabetes.49.12.1967, lire en ligne)
(en) A.H. Romano et T. Conway, « Evolution of carbohydrate metabolic pathways », Research in Microbiology, vol. 147, nos 6-7, juillet-septembre 1996, p. 448-455 (PMID 9084754, DOI 10.1016/0923-2508(96)83998-2, lire en ligne)
(en) Arthur L. Koch, « How Did Bacteria Come to Be? », Advances in Microbial Physiology, vol. 40, 1998, p. 353-399 (PMID 9889982, DOI 10.1016/S0065-2911(08)60135-6, lire en ligne)
(en) Christos Ouzounis et Nikos Kyrpides, « The emergence of major cellular processes in evolution », FEBS Letters, vol. 390, no 2, 22 juillet 1996, p. 119-123 (PMID 8706840, DOI 10.1016/0014-5793(96)00631-X, lire en ligne)
(en) Thomas Cavalier-Smith, « Rooting the tree of life by transition analyses », Biology Direct, vol. 1, 11 juillet 2006, p. 19 (PMID 16834776, PMCID 1586193, DOI 10.1186/1745-6150-1-19, lire en ligne)
(en) Gustavo Caetano-Anollés, Hee Shin Kim et Jay E. Mittenthal, « The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture », Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no 22, 29 mai 2007, p. 9358-9363 (PMID 17517598, DOI 10.1073/pnas.0701214104, lire en ligne)
(en) Steffen Schmidt, Shamil Sunyaev, Peer Bork et Thomas Dandekar, « Metabolites: a helping hand for pathway evolution? », Trends in Biochemical Sciences, vol. 28, no 6, juin 2003, p. 336-341 (PMID 12826406, DOI 10.1016/S0968-0004(03)00114-2, lire en ligne)
(en) Rui Alves, Raphael A.G Chaleil et Michael J.E Sternberg, « Evolution of Enzymes in Metabolism: A Network Perspective », Journal of Molecular Biology, vol. 320, no 4, 19 juillet 2002, p. 751-770 (PMID 12095253, DOI 10.1016/S0022-2836(02)00546-6, lire en ligne)
(en) Hee Shin Kim, Jay E Mittenthal et Gustavo Caetano-Anollés, « MANET: tracing evolution of protein architecture in metabolic networks », BMC Bioinformatics, vol. 7, 2006, p. 351 (PMID 16854231, DOI 10.1186/1471-2105-7-351, lire en ligne)
(en) Sarah A. Teichmann, Stuart C.G. Rison, Janet M. Thornton, Monica Riley, Julian Gough et Cyrus Chothia, « Small-molecule metabolism: an enzyme mosaic », Trends in Biotechnology, vol. 19, no 12, décembre 2001, p. 482-486 (PMID 11711174, DOI 10.1016/S0167-7799(01)01813-3, lire en ligne)
(en) Victor Spirin, Mikhail S. Gelfand, Andrey A. Mironov et Leonid A. Mirny, « A metabolic network in the evolutionary context: Multiscale structure and modularity », Proceedings of the National Academy of Sciences of the United States of America, vol. 103, no 23, 6 juin 2006, p. 8774-8779 (PMID 16731630, PMCID 1482654, DOI 10.1073/pnas.0510258103, lire en ligne)
(en) Jennifer J Wernegreen, « For better or worse: genomic consequences of intracellular mutualism and parasitism », Current Opinion in Genetics & Development, vol. 15, no 6, décembre 2005, p. 572-583 (PMID 16230003, DOI 10.1016/j.gde.2005.09.013, lire en ligne)
(en) Csaba Pál, Balázs Papp, Martin J. Lercher, Péter Csermely, Stephen G. Oliver et Laurence D. Hurst, « Chance and necessity in the evolution of minimal metabolic networks », Nature, vol. 440, 30 mars 2006, p. 667-670 (PMID 16572170, DOI 10.1038/nature04568, Bibcode 2006Natur.440..667P, lire en ligne)
(en) U. von Stockar et J.-S. Liu, « Does microbial life always feed on negative entropy? Thermodynamic analysis of microbial growth », Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1412, no 3, 4 août 1999, p. 191-211 (PMID 10482783, DOI 10.1016/S0005-2728(99)00065-1, lire en ligne)
(en) Y. Demirel et S.I. Sandler, « Thermodynamics and bioenergetics », Biophysical Chemistry, vol. 97, nos 2-3, 19 juin 2002, p. 87-111 (PMID 12050002, DOI 10.1016/S0301-4622(02)00069-8, lire en ligne)
Gillooly JF, Brown JH, West GB, Savage VM, Charnov EL (2001) Effects of size and temperature on metabolic rate. Science, 293, 2248–2251.
Clarke A, Johnston N (1999) Scaling of metabolic rate with body mass and temperature in teleost fish . Journal of Animal Ecology, 68, 893–905.
Clarke A (2004) Is there a universal temperature dependence of metabolism ? Functional Ecology, 18, 252–256.
S.V. Kumar et P.A. Wigge, H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis, Cell, vol. 140, pp. 136-147, 2010
Brève du journal Pour la Science (par Jean-Jacques Perrier, 2010/01/25)
(en) J. K. Nicholson, J. C. Lindon et E. Holmes, « 'Metabonomics': understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data », Xenobiotica, vol. 29, no 11, novembre 1999, p. 1181-1189 (PMID 10598751, DOI 10.1080/004982599238047, lire en ligne)
Fischer-Kowalski, M. (2011) Analyzing sustainability transitions as shifts between sociometabolic regimes. In: Environmental Innovation and Societal Transitions
Fischer-Kowalski, M. and H. Haberl (2007) Socioecological transitions and global change: Trajectories of Social Metabolism and Land Use. Edward Elgar, Cheltenham, UK and Northhampton, USA.
Eisenmenger, N. and Giljum, S. (2006) “Evidence from Societal Metabolism Studies for Ecological Unequal Trade” ; In: A. Hornborg and C.L. Crumley (eds.). The World System and the Earth System. Global Socio-Environmental Change and Sustainability since the Neolithic. Left Coast Press Inc.: Walnut Creek, California. 288-302.
Annexes |
Articles connexes |
- Anabolisme
- Catabolisme
- Métabolisme aérobie
- Métabolisme anaérobie
- Métabolisme de base
- Métabolite
- Métabolite secondaire
- Cachexie
- Cachexie cancéreuse
- Régime et gastronomie
- Biologie
- Sucres
- Lipides
Bibliographie |
- Brown JH, Gillooly JF, Allen AP, Savage VM, West GB (2004) Toward a metabolic theory of ecology. Ecology, 85, 1771–1789.
Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’ : understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999 ; 29 : 1181-9.
Liens externes |
Les principales voies du métabolisme : un cours de Biochimie sur wikilivres
- Interactive flow chart of the major metabolic pathways (engl.)
Le métabolisme interactif, Université P& M Curie- « métabolisme »
 Portail de la biochimie
Portail de la biochimie  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire  Portail de la chimie
Portail de la chimie


